For brevity’s sake, I’ll focus on trials about newly diagnosed MM and myeloma at first relapse. Here’s my take on how to interpret those studies in light of broader evidence, what I view as their key limitations, and how what came out of ASCO 2024 changes my approach.
The Return of Belantamab
Belantamab, a BCMA targeting antibody-drug conjugate, previously had shown a response rate of 34% in a single-arm, heavily pretreated population, albeit with modest progression free survival (PFS), only to fail its confirmatory randomized study against pomalidomide/dexamethasone. Given the ocular toxicity associated with belantamab, many — including myself — had written off this drug (save in exceptional/unique circumstances), especially with the rise of novel immunotherapies targeting BCMA, such as chimeric antigen receptor (CAR T-cell) therapy and bispecific antibodies.
 Huntsman Cancer Institute
Huntsman Cancer Institute
Manni Mohyuddin, MD
However, this year at ASCO, two key randomized trials were presented with concurrent publications, a trial of belantamab/bortezomib/dexamethasone versus daratumumab/bortezomib/dexamethasone (DVd) (DREAMM-7), and a trial of belantamab/pomalidomide/dexamethasone versus bortezomib/pomalidomide/dexamethasone (DREAMM-8). Both trials evaluated patients with myeloma who had relapsed disease and had received at least one prior line of therapy.
In both trials, the belantamab triplet beat the other triplets for the endpoint of PFS (median PFS 36.6 vs 13 months for DREAMM-7, and 12 months PFS 71% vs 51% for DREAMM-8). We must commend the bold three-versus-three design and a convincing result.
What are the caveats? Some censoring of information happened in DREAMM-7, which helped make the intervention arm look better than reality and the control arm look even worse than reality. To illustrate this point: the control arm of DVd (PFS 13 months) underperformed, compared to the CASTOR trial, where DVd led to a PFS of 16.7 months. The drug remains toxic, with high rates of keratopathy and vision problems in its current dosing schema. (Perhaps the future lies in less frequent dosing.) This toxicity is almost always reversible, but it is a huge problem to deal with, and our current quality-of-life instruments fail miserably at capturing this.
Furthermore, DVd is now emerging as perhaps the weakest daratumumab triplet that exists. Almost all patients in this trial had disease sensitivity to lenalidomide, and daratumumab/lenalidomide/dexamethasone (PFS of 45 months in the POLLUX trial) is unequivocally easier to use and handle (in my opinion) than this belantamab triplet--which is quite literally “an eyesore.” Would belantamab-based triplets beat dara/len/dex for patients with lenalidomide sensitive disease? Or, for that matter, would belantamab combos beat anti-CD38+carfilzomib+dex combinations, or cilta-cel (which is also now approved for first relapse)?
How do I foresee the future of belantamab? Despite these unequivocally positive results, I am not enthused about using it for most patients at first relapse. When trials for bispecifics at first relapse read out, my enthusiasm will likely wane even more. Still, it is useful to have belantamab in the armamentarium. For some patients perceived to be at very high risk of infection, belantamab-based triplets may indeed prove to be a better option than bispecifics. However, I suspect that with better dosing strategies for bispecifics, perhaps even that trend may be mitigated. Since we do not yet have bispecifics available in this line, my suggested algorithm for first relapse is as follows:
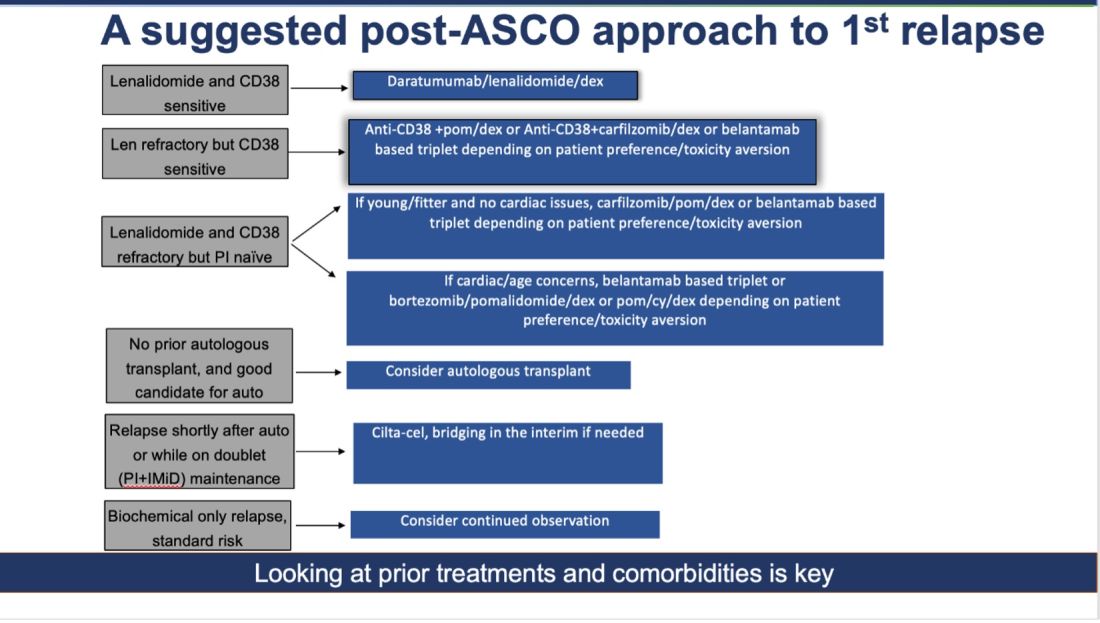 Courtesy Dr. Mohyuddin
Courtesy Dr. Mohyuddin