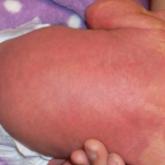
Presentation The characteristic clinical features of SLS begin to develop during the intranatal period and infancy.1,6 Pathologic skin involvement can be detected as early as week 23 of gestation. Preterm births associated with SLS have commonly been described.3 Ichthyosis often is evident at birth, but collodion membrane is uncommon. Severe pruritus is a marked feature unlike most other types of ichthyosis. The ichthyosis often is generalized with prominent involvement of the flexural areas and nape of the neck, varying from fine furfuraceous to larger lamellarlike scales. Velvety orange or brown lichenification often is a predominant feature in the flexures of the arms, legs, neck, and mid abdomen. Mental retardation, developmental delay, and spasticity usually become apparent at 1 to 2 years of age and subsequently are nonprogressive.6,7 However, patients rarely have been described with normal intellectual functioning.7 Spasticity often is more severe in the lower limbs and may lead to contractures, kyphoscoliosis, hip dislocation, and short stature. Delayed speech and dysarthria are common. Parafoveal glistening white dots on the retina are a pathognomonic feature and typically appear in the first 2 years of life; however, they are seen in approximately 30% of patients and increase slightly in number with age.6,8 There may be associated decreased visual acuity, photophobia, myopia, and astigmatism. Other clinical features include enamel hypoplasia, metaphyseal dysplasia, and epilepsy.1,6
Gene Mutations Sjögren-Larsson syndrome is caused by mutation in thealdehyde dehydrogenase 3 family member A2 gene, ALDH3A2 (17p11.2), which codes for FALDH.1,6,7 The ALDH3A2 gene is 11 exons long and gives rise to 2 protein isoforms that differ in their carboxy-terminal domains; the major isoform, composed of 485 amino acids, localizes to the endoplasmic reticulum. The minor protein isoform (FALDHv) is composed of 508 amino acids, possesses a longer carboxy-terminal, and appears to be targeted to the peroxisome. Several mutations have been reported throughout the ALDH3A2 gene, including missense mutations (most common [38% of cases of SLS6]), deletions, insertions, splicing errors, and complex rearrangements. Although several of these mutations are private, several common mutations may be indicative of founder effects (ie, shared ancestry), consanguinity, or recurrent mutational events (mutation hotspots).6,7 Despite the wide spectrum of mutations, there is very little phenotypic variation, with consistently severe cutaneous and neurological involvement occurring in a majority of patients.7 However, Lossos et al9 described remarkable phenotypic variation in 6 siblings of an Arab family and suggested that additional unknown genetic or environmental factors may compensate for the biochemical defect.
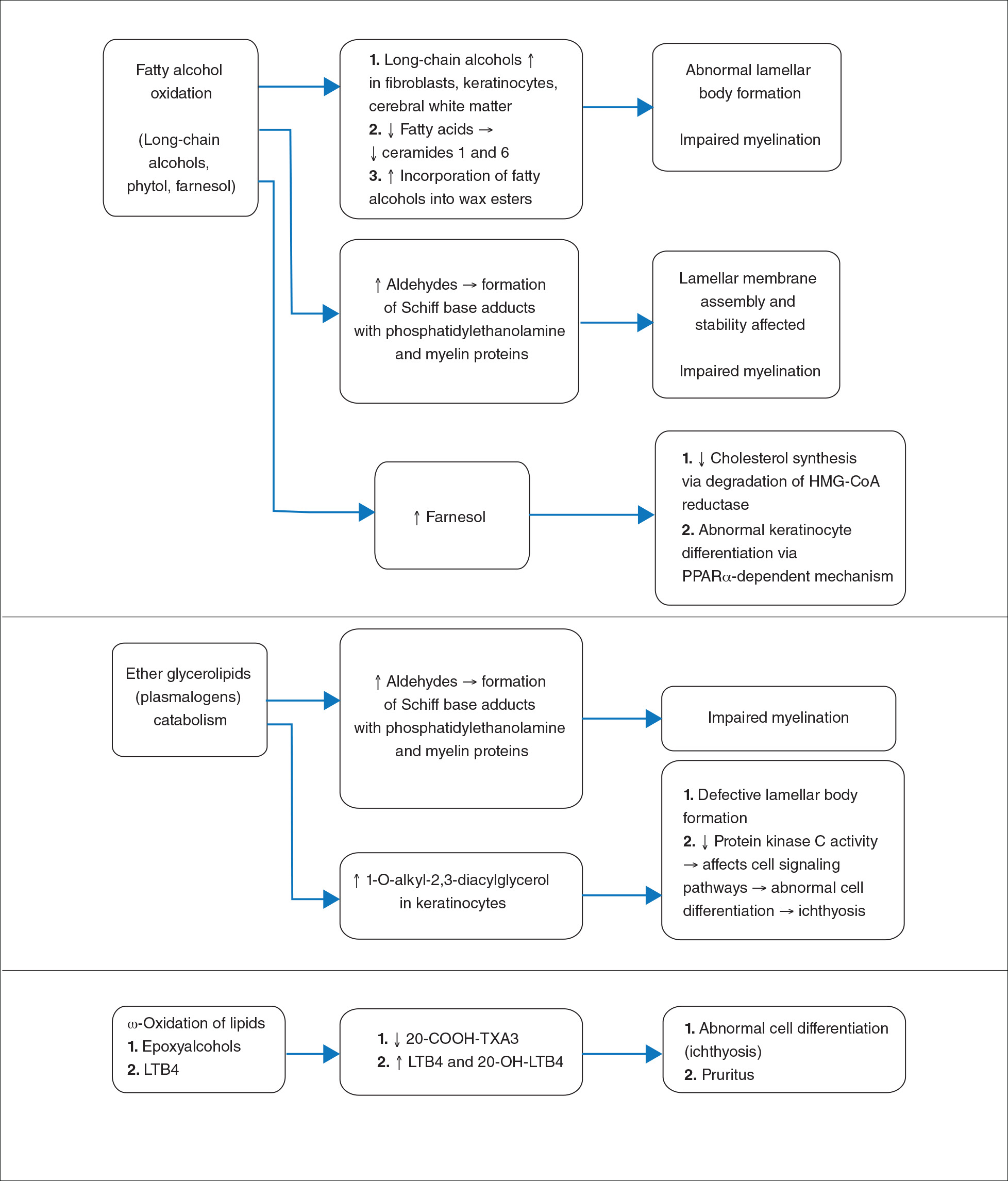
Lipid Metabolism Fatty aldehyde dehydrogenase is expressed in almost all cells and tissues and catalyzes the oxidation of fatty aldehydes to fatty acids (eFigure 1). It also is a part of the fatty alcohol:NAD oxidoreductase (FAO) enzyme complex, which catalyzes fatty alcohol oxidation to fatty acid. Fatty aldehyde dehydrogenase deficiency leads to accumulation of long-chain alcohols (eg, hexadecanol, octadecanol, octadecenol) and diversion of fatty alcohol into alternate biosynthetic pathways such as wax esters and 1-O-alkyl-2,3-diacylglycerol.10 Other lipids that are increased are illustrated in eFigure 2. Accumulation of these lipids, toxic effects of abnormal lipids (especially fatty aldehydes and Schiff base protein-lipid adducts), and lack of essential lipids (eg, polyunsaturated fatty acids, ceramides 1 and 6, triglycerides) are responsible for the classical cutaneous, neurologic, and ophthalmologic features of SLS.
eFigure 1. Role of fatty aldehyde dehydrogenase (FALDH) in lipid metabolism in Sjögren-Larsson syndrome. Fatty aldehyde dehydrogenase is responsible for oxidation of fatty aldehydes derived from long-chain alcohols, branched alcohols, isoprenoid alcohols, and ether glycerolipids. Fatty aldehyde dehydrogenase also is necessary for ω-oxidation of leukotriene B4 (LTB4) and epoxyalcohols (trioxillin A3). Lipid metabolites shown in italics are increased in Sjögren-Larsson syndrome.
Histopathology The epidermal permeability barrier is critically dependent on the appropriate lipid composition of the multilamellar stratum corneum intercellular membranes, an equimolar ratio of cholesterol, ceramides, and fatty acids. Histopathology of the skin in SLS generally shows hyperkeratosis, papillomatosis, acanthosis, and a mildly thickened granular layer. Ultrastructural studies of the skin reveal misshapen/empty lamellar bodies, abnormal cytoplasmic lamellar inclusions in the granular keratinocytes, lipid droplets in the stratum corneum with decreased lamellar bilayers, and lamellar/nonlamellar phase separation in the stratum corneum interstitium.11 These findings indicate that lipid metabolism dysfunction in SLS results in marked impairment in formation and secretion of lamellar bodies in the epidermis and consequent disorganization of the stratum corneum lamellar membranes. The resulting disruption of the skin barrier function leads to increased transepidermal water loss, resulting in ichthyosis.11,12 Another proposed mechanism for ichthyosis in SLS is disruption of the normal epidermal differentiation resulting from abnormal lipid metabolites (eFigure 2). Also, increased leukotriene B4 (LTB4) and 20-hydroxy-leukotriene B4 (20-OH-LTB4)(eFigure 1) may be responsible for the considerable pruritus seen in SLS.10
Neurologic Findings Neurologic changes in SLS result from delayed and deficient myelination. Neuropathological studies have shown ballooning of myelin sheaths, extensive loss of myelin, axonal damage, and astrogliosis. The presence of lipoid material positive for periodic acid–Schiff that stains light rather than dark pink, dense distribution of round/ellipsoid bodies in the white matter of the cerebrum and brainstem positive for periodic acid–Schiff, and proliferation of perivascular macrophages containing lipofuscinlike pigments also have been described.13 Possibly, in the absence of FALDH, metabolism of plasmalogens (a major component of myelin) results in increased fatty aldehydes, which are either diverted to fatty alcohols or form adducts with phosphatidylethanolamine and myelin basic proteins (eFigure 1). Magnetic resonance imaging of the brain usually shows hypomyelination involving the periventricular white matter extending from the frontal to the occipital area.7,14 Mild ventricular enlargement may be an additional feature.14
A useful application of MRI is the proton magnetic resonance spectroscopy, which quantifies the brain metabolites noninvasively, displaying them as a spectrum on a graph. The spectrum comprises a set of resonances/peaks distributed along an x-axis. The resonances of these metabolites are obtained after suppressing the large signals from water protons. Proton magnetic resonance spectroscopy of the normal brain shows 3 prominent peaks: (1) N-acetylaspartate (NAA) at 2.02 ppm, (2) creatine at 3.02 ppm, and (3) choline at 3.22 ppm. In SLS, cerebral proton MRI spectroscopy reveals a characteristic abnormal, prominent, and narrow lipid peak at 1.3 ppm (corresponding to hexadecanol and octadecanol) and may offer a quantitative parameter for monitoring the effects of therapeutic interventions.7,14,15 The most intense lipid peaks are located in the periventricular regions in the anterior and posterior trigones. An abnormal but much smaller peak may be seen at 0.8 to 0.9 ppm, corresponding to phytol.14 Gradual emergence of these changes occurs in the first 2 years of life and then remains stable.15 Proton magnetic resonance spectroscopy also can be used for screening of SLS heterozygotes.16 Lipid peaks have been described in other disorders of lipid metabolism, but they are less intense, broader, and disappear on longer echo time sequences.14
Besides the characteristic parafoveal glistening white dots the retina, optical coherence tomography shows focal hyperreflectivitity in the perifoveal ganglion cell layer and inner plexiform layer of the retina as well as cystoid foveal degeneration.17 The intraretinal deposition of lipid metabolites probably leads to Müller cell degeneration with subsequent formation of cystoid spaces and atrophic changes in the fovea.
Measurement of FALDH or FAO activity in cultured skin fibroblasts and leukocytes using flurometric or gas chromatography mass spectrometry assays is a reliable biochemical test in cases of SLS as well as in heterozygotes.17 A decrease in FALDH/FAO activity also can be demonstrated by histochemical staining in skin biopsy.11 Pathologic urinary excretion of LTB4 and 20-OH-LTB4 also is a biochemical marker of SLS. Mutation analysis for a specific gene defect is diagnostic in cases of SLS as well as in heterozygotes. Prenatal diagnosis of SLS is possible by assessing FALDH activity or gene defects in cultured chorionic villus fibroblasts and amniocytes.18,19
Differential Diagnosis The differential diagnosis of SLS includes congenital ichthyosiform erythroderma with neurological signs (Tay syndrome, Conradi-Hünermann-Happle syndrome) and neurocutaneous disorders such as neutral lipid storage disease and multiple sulfatase deficiency; however, the nature of the ichthyosis, presence of spastic diplegia/tetraplegia, characteristic parafoveal glistening white dots on the retina, and MRI and proton magnetic resonance spectroscopy findings help to easily differentiate SLS from these disorders.
Treatment Treatment of SLS mainly is palliative. Ichthyosis can be treated with topical keratolytics, emollients, calcipotriol, and oral retinoids (acitretin).6 Zileuton, a 5-lipoxygenase inhibitor, inhibits synthesis of LTB4 and cysteinyl leukotrienes, thereby reducing the severity of pruritus and also has been shown to improve the speed of information processing.18 Similarly, montelukast, a leuko-triene antagonist, is helpful in relieving the agonizing pruritus.19 Experimental studies have shown that bezafibrate, a peroxisome proliferator-activated receptor α agonist, induces FALDH activity in fibroblasts of SLS patients that still have some residual FALDH activity, but further research is required to determine whether SLS patients could benefit from treatment.20 Physiotherapy helps in relieving the spasticity to some extent, such as in our case. Dietary intervention with reduced fat intake (up to 30% of total daily calorific requirement) and supplementation with omega-3 and omega-6 fatty acids has shown variable results in anecdotal reports.21-23 Gene therapy using recombinant adeno-associated virus 2 vectors to restore FALDH has been projected as a future treatment option.24 Despite lack of effective treatment options, most patients of SLS survive well into adulthood.
Conclusion
Because ichthyosis is one of the earliest and prominent symptoms of SLS, a dermatologist can play an important role in early diagnosis. Any child with the classical pattern of ichthyosis should be thoroughly examined for early neurologic signs and investigated to rule out SLS. Proton magnetic resonance spectroscopy serves as a useful adjunct in the diagnosis of SLS by confirming the accumulation of abnormal lipids in the periventricular white matter, especially when specific enzyme analysis and genetic analysis are not available in resource-restricted settings.